|
Abstract: |
Introduction
The oral administration of anticancer drugs is becoming increasingly popular. Around 20–25% of anticancer drugs are oral forms, including targeted therapies now established for indications including chronic myeloid leukaemia, gastrointestinal stromal tumour or renal cancer [1].
This route is beneficial for several reasons, including pharmacoeconomics (reduced production and staffing costs compared to the delivery of parenteral medications), patients’ preferences (autonomy, limited or no hospitalization, absence of venous access, improved quality of life especially for those undergoing palliative treatment) as well as for pharmacokinetics, pharmacodynamics and biopharmaceutical aspects. Indeed, for drugs with similar pharmacokinetic profiles following oral and parenteral administration, such as hydroxycarbamide and vinorelbine, oral formulations are equally as effective as parenteral [2].
Oral administration of drugs enables lower concentrations of cytotoxic agents to be administered but with a more prolonged exposure in the body compared to the systemic route. This may be more effective particularly for anticancer agents that are cell cycle phase-dependent such as antimetabolites. Indeed, chronic administration leads to a steady state of the plasma concentration which cannot be obtained by intermittent IV administration [2, 3]. Therefore, oral administration can mimic continuous infusion such as for capecitabine as compared to 5FU.
Some prodrugs require metabolic activation. Through the oral route, passage through the intestinal and subsequent metabolism by the liver will increase the amount of active molecules reaching the systemic circulation. This is therefore the preferred route for several prodrugs including capecitabine, altretamine, etoposide phosphate, estramustine natrium phosphate [4].
The oral route is also preferred for drugs that are poorly soluble in water or in biocompatible solvents, such as altretamine, finasteride, chlorambucil, melphalan and tretinoin.
Despite these advantages, the oral route may also present difficulties. For example, food–drug interactions may affect the bioavailability of some drug formulations, and hence new formulations need to be assessed in clinical studies if they are to be given orally. Furthermore, the oral route may not be optimal for anticancer drugs that do not need prolonged exposure, such as those administered by IV bolus once every three weeks. A drug exhibiting too large a variation in bioavailability is not a good candidate for oral administration, particularly if it has a very low therapeutic index, as occurs with anticancer agents such as anthracyclines or vinca alkaloids. Finally, side effects such as local irritation, nausea, emesis or diarrhoea following chronic oral administration can seriously jeopardize patient compliance, especially after daily home use. This in turn can alter the overall dose-intensity and clinical results [4].
Food can significantly affect the pharmacokinetics of orally administered drugs [5–7] but this can be managed by administering anticancer drugs at regular times in relation to food intake. As pharmacists have the potential to play an increasingly important role in optimising the oral administration of anticancer drugs, it is important to recall the factors influencing their oral bioavailability and the methods that can predict this.
General factors affecting oral drug bioavailability
Bioavailability is defined as the fraction of an active substance that reaches the central compartment, taking into account the speed with which this compartment is fulfilled [8]. Numerous factors can affect oral bioavailability: either extrinsic, i.e. related to the physicochemical properties of a molecule, its formulation or drug or food interactions; or intrinsic, related to the individual patient’s physiology [6].
Extrinsic factors
The physicochemical properties of a molecule as well as its solubility and membrane permeability (correlated to the log Poctanol/water) strongly influence its oral absorption. A classical study by Amidon et al [9] has led to a biopharmaceutical classification system (BCS) which roughly divides any drug into four classes according to its solubility and permeability parameters, see Figure 1. This classification can not only predict the oral absorption of a molecule but also evaluates the influence of food-related physiological change on absorption [6, 9, 10]. Class I indicates no significant food effects; Class II refers to enhanced solubility and absorption of a drug when taken with food—except for certain formulations containing acids or weak bases with a low pK, which tend to precipitate as pH increases following food intake; Class III indicates a negative effect of food; Class IV indicates drugs for which effects are difficult to predict [9, 10].
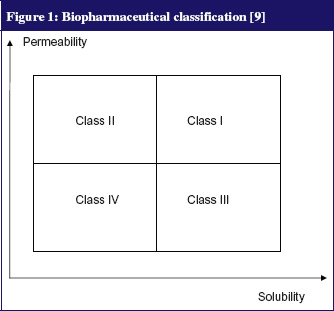
The dosage and formulation also influence the rate of drug release and its availability in the intestine for absorption. For example, the speed of disintegration of a solid oral form may depend heavily on the environment of the gastrointestinal tract (pH, gastric fluid volume). Finally, the oral bioavailability of drugs can be affected greatly by the concomitant use of another drug or food, which can either alter a drug’s physicochemical state (chelation, solubilisation) and/or its physiological state through changing the gastrointestinal environment (gastric emptying, pH).
Intrinsic factors
These are mostly physiological factors that cause variability in oral bioavailability over time within the same person or between individuals. These include membrane permeability, electrical resistance [11], the number of pores within tight junctions [12], the activity of transporter proteins such as the solute-carrier gene (SLC) transporter superfamily [13] and the ATP-binding cassette (ABC) transporter superfamily [14], the intestinal first-pass effect of enzymes in enterocytes, namely cytochromes P450 (especially CYP3A4) and conjugating enzymes [15], and bile and digestive secretions (including enzymes derived from the pancreas, bacterial flora or the small intestine). Bile acids may form complexes with, and lead to the sequestration of some drugs in micelles, thus decreasing their concentration and reducing their luminal absorption [16]. Conversely, bile acids solubilise lipophilic drugs and increase their absorption [6]. The rate of gastric emptying also affects the rate and extent of metabolism and intestinal absorption of drugs by determining the time taken to reach the preferential site of absorption, the proximal intestine [16]. The motility and intestinal transit time also determine how long drugs remain at the absorption sites, while the pH of the intestinal fluid mainly affects the absorption of weak acids and bases. With regard to pH, it is worth remembering that only the unionized form of a drug can be absorbed by simple diffusion; weakly acidic drugs dissolve more quickly at high pH, while weak bases prefer lower pH. In addition, the volume of the gastric contents can lead to a saturation effect that limits uptake of poorly soluble substances. Finally, active intestinal blood flow (500 mL/mn) is directly related to the metabolic demand of intestinal cells and their functional activity. Thus, an increased intestinal blood flow will reduce the intestinal first-pass metabolism of a drug [17].
Finally, the influence of food–drug interactions is of paramount practical importance given that most of new targeted therapies such as tyrosine kinase inhibitors are highly lipophilic and heavily metabolized by cytochromes.
Specific effects of foods on cancer drug bioavailability
Food can interact with oral anticancer drugs either directly, by chelation, complexation through ion-pair formation or physical obstruction at the absorption site, or indirectly, through the impact of food intake on physiological parameters. Physiological parameters of the gastrointestinal tract include slowing of gastric emptying, increased transit times, increased pH, volume and viscosity of gastrointestinal contents, increased volume of secretions including digestive bile fluid, increased hepatic blood flow and lymphatic transport. On a metabolic level, some foods can induce or inhibit intestinal metabolism (including cytochromes) and intestinal transport, both influx and efflux [4–7, 10].
Food effects can vary between causing delay, reduction, increase or acceleration of absorption, and having no detectable effect [4, 5, 18]. These interactions are reflected by changes in Cmax (maximal plasma concentration, referred to as peak), Tmax (time to reach Cmax) and/or the area under the plasma curve (AUCo-t) or extrapolated to infinity AUCo-8.
These interactions and subsequent recommendations for the use of oral cancer drugs are presented in Table 1.
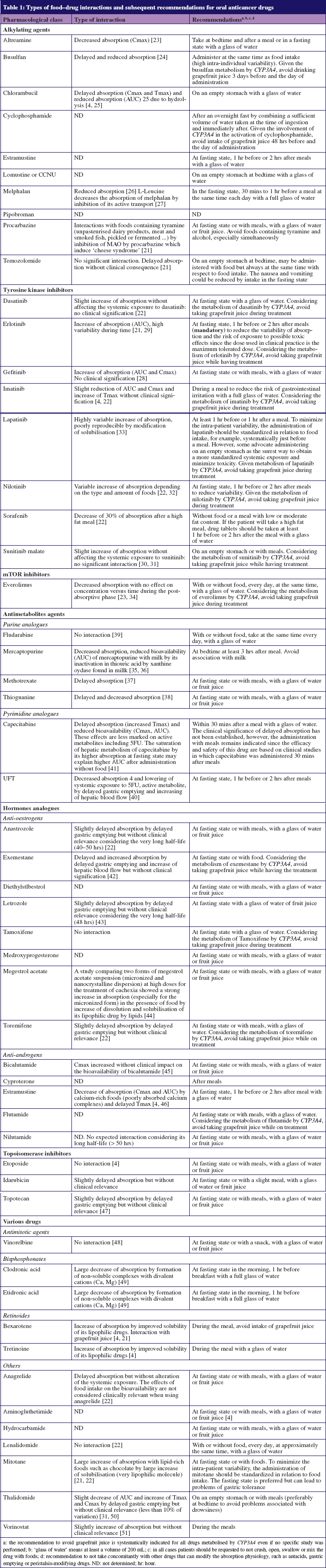
Delayed absorption
Delayed absorption is usually manifested by a decrease in Cmax, and extension of Tmax but without significant effect on the amount of drug absorbed (no modification of AUC).
Such interactions may be due to the slowing of gastric emptying and delayed presentation in the small intestine where the drug is normally absorbed.
This interaction seems to occur with molecules assigned to BCS Class 1 (as mentioned previously) which have good solubility and permeability, provided that they are stable in the gastric environment [4–6]. Delayed absorption may cause a delay in therapeutic activity. Generally, however, this is not clinically significant, given that oral cancer agents are often used for chronic long-term administration, for which a delay in absorption may be beneficial as a means to avoid certain side effects or acute toxicity [5].
Decreased absorption
Decreased absorption occurs following modification in AUC, which reflects the amount absorbed, and a concomitant decrease of Cmax. Mechanisms that decrease absorption are: 1) drug instability in gastrointestinal fluids, as with chlorambucil which undergoes more hydrolysis with longer time in the gastrointestinal tract; 2) physical or chemical binding between drugs and certain food components, such as estramustine phosphate sodium which forms a less absorbable complex with calcium in the presence of milk or other calcium-rich foods; 3) solid foods limiting the number of molecules that reach the intestinal wall for absorption; 4) an increase in the first-pass effect following the action of enzyme-inducing compounds present in particular food proteins; 5) effects on drug transport by certain amino acids such as L-leucine and L-glutamic acid, which interfere, for example, with the transport of melphalan probably by competitive inhibition but do not influence the transport of passively absorbed drugs such as chlorambucil [4, 5].
These interactions preferentially affect compounds in BCS Class III (high solubility/low permeability) whose permeability is the limiting factor for absorption.
Accelerated or increased absorption
This occurs when there is an increase in the AUC accompanied by an increase in Cmax and sometimes a decrease in Tmax. Several mechanisms may be involved: 1) the increased secretion of bile salts which can improve the solubility of lipophilic drugs by micelle formation and for hydrophilic drugs may form better absorbed ion pairs; 2) prolonged residence time in the gastrointestinal tract due to an increased volume of gut contents (either by drink or by the digestive secretions); 3) the binding of lipophilic drugs to dietary lipoproteins to increase the solubility of poorly soluble drugs; 4) a decrease in the first-pass effect linked to an increase in lymphatic transport especially after a high-fat meal or an increased splanchnic flow passing from 1,200 mL/min/m2 to 1,600 mL/min/m2 after a high-protein meal, resulting in a decreased transit time through the liver, thus limiting the hepatic first-pass effect and increasing the systemic concentration of non-metabolized drug [16].
These interactions are mainly observed for BCS class II drugs for which solubility limits absorption. For example, a high fat meal increases the solubilisation of lapatinib, exemestrane, etretinate, fenretinide and tretinoin.
Absorption unaffected
Some drugs are insensitive to changes in the gastrointestinal tract that occur after food intake. These are usually rapidly and completely absorbed, as in the case of finasteride, or well absorbed both in the small intestine and colon as in the case of bicalutamide.
These drugs therefore offer greater flexibility over the relative timing of meals, which may increase patient compliance. It may also be possible to divide the dose into fractions to improve efficiency, as in the case of etoposide, and security, as occurs with vinorelbine where lower doses result in the reduction of gastrointestinal toxicity [4–6].
Methods for evaluating the effect of foods on oral cancer bioavailability
FDA has recognized that foods can affect the pharmacokinetics of certain drugs. In December 2002, FDA issued instructions to manufacturers on how to investigate the impact of food on oral bioavailability and bioequivalence: ‘Guidance for Industry: food-effect bioavailability and fed bioequivalence studies’ [7]. To study the impact of food intake on the oral bioavailability of drugs, FDA recommends performing a randomized, crossover, single dose study, with two treatments in two periods: during fasting state and after a test meal with an adequate washout period between the two regimens. The study requires, if drug safety permits, a sufficient number (at least 12) of healthy volunteers. The test meal is eaten in a way that generates the greatest impact possible on the gastrointestinal physiology in order to allocate the maximum bioavailability of the drug studied. This usually means a high-fat meal (with fat comprising at least 50% of the calories consumed) and high-calorie (800 to 1,000 kcal in total). Given anticancer drugs to volunteers for such a study is not permitted for ethical reasons.
However, the prediction of possible food–drug related effects on oral anticancer drugs is critical, especially for drugs used chronically such as tyrosine kinase inhibitors, where such interactions could lead to side effects, poor compliance and, ultimately, reduced efficacy [19]. Indeed, food interaction can lead to an increasing absorption with subsequent higher plasma levels inducing more side effects which can decrease compliance.
Several experimental, non-clinical methods have been developed for predicting the possible effects of food on drug pharmacokinetics. These include in vitro methods based on certain physicochemical parameters related to the active substance, such as dose/solubility and LogP, and the BCS classification based on solubility parameters and permeability. These in vitro methods have the advantage of using easily measurable parameters and enable screening for food effects to be performed early in drug development. However, they do not provide a quantitative measure of changes nor explain their mechanisms [19]. More recently, computer software such as GastroPlus has been used to simulate the effect of food on a substance through physiologically based pharmacokinetic models using data such as permeability, solubility, metabolism and distribution; and physiological parameters [19, 20].
Dissolution tests that artificially recreate the gastrointestinal environment during fasting or after food intake (by addition of bile salts and lecithin) also appear to be predictive about the impact of food on bioavailability. This method is simple to implement and provides good results for BCS class II substances, but needs optimising and standardising in the compositions of media used. Where possible food–drug interactions are suspected through the results of such in vitro methods, it is essential to design complementary animal studies, especially for anticancer drugs intended for chronic use. For in vivo methods, dogs appear to be the most suitable animal model for studying the effect of food on bioavailability although there are some physiological differences from humans in terms of gastric pH, gastric emptying and intestinal transit time. Other frequently used models are monkeys and guinea pigs. Meanwhile, some teams have attempted to evaluate drug solubility in aspirated human or dog intestinal fluids, taken either before or after a meal [19]. In summary, if food interactions are likely and for drugs intended for chronic use (targeted therapies, hormonal therapy), these should be evaluated through clinical studies. The study of dietary interactions should ideally be conducted during phase I clinical trials, but this is difficult with cytotoxic molecules. Thus, relevant information on these interactions should be obtained during the phase II or III through the implementation of prospective population-based pharmacokinetic studies.
Author for correspondence
Professor Alain Astier, PharmD, PhD
Head of Department
Department of Pharmacy
CNRS-UMR 7054
Henri Mondor University Hospitals
51 avenue du Maréchal de Lattre de Tassigny
FR-94010 Créteil, France
Co-author
Amal Yacoub
Pharmacy Student
References
1. Ranchon F, Bouret C, Charpiat B, Leboucher G. Sécurisation de l’emploi des chimiothérapies anticancéreuses administrables par voie orale. Le Pharmacien hospitalier. 2009;44(1):36-44. French.
2. Greco FA. Evolving role of oral chemotherapy for the treatment of patients with neoplasms. Oncology (Williston Park). 1998;12(3 Suppl 4):43-50.
3. O’Neill VJ, Twelves CJ. Oral cancer treatment: developments in chemotherapy and beyond. Br J Cancer. 2002;87(9):933-7.
4. Singh BN, Malhotra BK. Effects of food on the clinical pharmacokinetics of anticancer agents: underlying mechanisms and implications for oral chemotherapy. Clin Pharmacokinet. 2004;43(15):1127-56.
5. Singh BN. Effects of food on clinical pharmacokinetics. Clin Pharmacokinet. 1999;37(3):213-55.
6. Fleisher D, Li C, Zhou Y, Paol H, Karim A. Drug, meal and formulation interactions influencing drug absorption after oral administration. Clinical implications. Clin Pharmacokinet 1999;36(3):233-54.
7. US Food and Drug Administration [homepage on the Internet]. Guidance for Industry: Food-effect bioavailability and fed bioequivalence studies [cited 2012 Sept 9]. Food and Drug Administration; Rockville, MD: 2002. Available from: www.fda.gov/eder/guidance/index.htm
8. Moulin M, Coqueret A. Pharmacologie. 2nd ed. Masson; 2002.
9. Amidon GL, Lennernas H, Shah VP, Crison JR. A theoretical basic for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res. 1995;12(3):413-20.
10. Custodio JM, Wu CY, Benet LZ. Predicting drug disposition, absorption/elimination/transporter interplay and the role of food on drug absorption. Adv Drug Deliv Rev. 2008;60(6):717-33.
11. Masaoka Y, Tanaka Y, Kataoka M, Sakuma S, Yamashita S. Site of drug absorption after oral administration : assessment of membrane permeability and luminal concentration of drugs in each segment of gastrointestinal tract. Eur J Pharm Sci. 2006;29(3-4):240-50.
12. Lacombe 0, Woodley J, Solleux C, Delbos JM, Boursier-Neyret C, Houin G. Localisation of drug permeability along the rat small intestine, using markers of the paracellular, transcellular and some transporters routes. Eur J Pharm Sci. 2004;23(4-5):385-91.
13. Sai Y, Tsuji A. Transporter-mediated drug delivery: recent progress and experimental approaches. Drug Discov Today. 2004;9(16):712-20.
14. Takano M, Yumoto R, Murakarni T. Expression and function of efflux drug transporters in the intestine. Pharmacol Ther. 2006;109(1-2):137-61.
15. Paine MF, Thummel KE. Role of intestinal cytochromes P450 in drug sr di position. In: Ortiz de Montellano PR, editor. Cytochrome P450: Structure, Mechanism and Biochemistry. 3rd ed. New York: Kluver Academic, Plenum Publishers; 2005. Chapter 12. p. 421-51.
16. DeSesso JM, Jacobson CF. Anatomical and physiological parameters affecting gastrointestinal absorption in humans and rats. Food Chem Toxicol. 2001;39(3):209-28.
17. Lin JH, Chiba M, Baillie TA. Is the role of the small intestine in first-pass metabolism overemphasized? Pharmacol Rev. 1999;51(2):135-58.
18. Yu LX, Straughn AB, Faustin PJ,Yang Y, Parekh A, Ciavarella AB, et al. The effect of food on the relative bioavailability of rapidly dissolving immediate-release solid oral products containing highly soluble drugs. Mol Pharm. 2004;1(5):357-62.
19. Lentz KA. Current methods for predicting human food effect. AAPS J, 2008;10(2):282-8.
20. Parrott N, Lukacova V, Fraczkiewicz G, Bolger MB. Predicting pharmacokinetics of drugs using physiologically based modeling—application to food effects. AAPS J. 2009;11(1):45-53.
21. BC Cancer Agency, Canada [homepage on the Internet]. BCCA cancer drug manual. [cited 2012 Oct 19]. Available from: www.bccancer.bc.ca/HPI/DrugDatabase
22. Dorosz PH, Vital Durand D, Le Jeune C, Guide pratique des médicaments. 31 st ed. Maloine: Paris; 2012. French.
23. Barker IK, Crawford SM, Fell AF. Determination of altretamine in human plasma with high-performance liquid chromatography. J Chromatogr B Biomed Appl. 1994;660(1):121-6.
24. Schuler U, Schroer S, Kühnle A, Blanz J, Mewes K, Kumbier I, et al. Busulfan pharmacokinetics in bone marrow transplant patients: is drug monitoring warranted? Bone Marrow Transplant. 1994;14(5):759-65.
25. Adair CG, Bridges JM, Desai ZR. Can food affect the bioavailability of chlorambucil in patients with haematological malignancies? Cancer Chemother Pharmacol. 1986;17(1):99-102.
26. Reece PA, Kotasek D, Morris RG, Dale BM, Sage RE. The effect of food on oral melphalan absorption. Cancer Chemother Pharmacol. 1986;16(2):194-7.
27. Reece PA, Dale BM, Morris RG, Kotasek D, Gee D, Rogerson S, et al. Effect of L-leucine on oral melphalan kinetics in patients. Cancer Chemother Pharmacol. 1987;20(3):256-8.
28. Swaisland HC, Smith RP, Laight A, Kerr DJ, Ranson M, Wilder-Smith CH, et al. Single-dose clinical pharmacokinetic studies of gefitinib. Clin Pharmacokinet. 2005;44(11):1165-77.
29. Ling J, Fettner S, Lum BL, Riek M, Rakhit A. Effect of food on the pharmacokinetics of erlotinib, an orally active epidermal growth factor receptor tyrosine-kinase inhibitor, in healthy individuals. Anticancer Drugs. 2008;19(2):209-16.
30. Bello CL, Sherman L, Zhou J, Verkh L, Smeraglia J, Mount J, et al. Effect of food on the pharmacokinetics of sunitinib malate (SUI 1248), a multi-targeted receptor tyrosine kinase inhibitor: results from a phase I study in healthy subjects. Anticancer Drugs. 2006;17(3):353-8.
31. Jiménez Torres NV, Romero Crespo I, Ballester Solaz M, Albert Mari A, Jiménez Arenas V. Antineoplastic oral agents and drug-nutrient interactions: a systematic review. Nutr Hosp. 2009;24(3):260-72.
32. Tanaka C, Yin OQ, Sethuraman V, Smith T, Wang Y, Grouss K, et al. Clinical pharmacokinetics of the BCR-ABL tyrosine kinase inhibitor nilotinib. Clin Pharmacol Ther. 2010;87(2):197-203.
33. Koch KM, Reddy NJ, Cohen RB, Lewis NI, Whitehead B, Mackay K, et al. Effects of food on the relative bioavailability of lapatinib in cancer patients. J Clin Oncol. 2009;27(8):1191-6.
34. Kovarik JM, Hartmann S, Figueiredo J, Rordorf C, Golor G, Lison A, et al. Effect of food on everolimus absorption: quantification in healthy subjects and a confirmatory screening in patients with renal transplants. Pharmacotherapy. 2002;22(2):154-9.
35. Burton NY, Bamett MJ, Aherne GW, Evans J, Douglas I, Lister TA. The effect of food on the oral administration of 6-mercaptopurine. Cancer Chemother Pharmacol. 1986;18(1):90-1.
36. Rivard GE, Lin KT, Leclerc JM, David M. Milk could decrease the bioavailability of 6-mercaptopurine. Am J Pediatr Hematol Oncol. 1989;11(4):402-6.
37. Pinkerton CR, Welshman SG, Glasgow JF, Bridges JM. Can food influence the absorption of methotrexate in children with acute lymphoblastic leukaemia? Lancet. 1980;1;2(8201):944-6.
38. Lancaster DL, Patel N, Lennard L, Lilleyman JS. 6-Thioguanine in children with acute lymphoblastic leukaemia: influence of food on parent drug pharmacokinetics and 6-thioguanine nucleotide concentrations. Br J Clin Pharmacol. 2001;51(6):531-9.
39. Oscier D, Orchard JA, Culligan D, Cunningham D, Johnson S, Parker A, et al. The bioavailability of oral fludarabine phosphate is unaffected by food. Hematol J. 2001;2(5):316-21.
40. Dan-de B, Ravandi F, Kaul S, Sonnichsen D, Ferreira I, Brooks D, et al. Effect of food on the oral bioavailability of UFT and Leucovorin in cancer patients. Clin Cancer Res. 2001;7(3):517-23.
41. Reigner B, Verweij J, Dirix L, Cassidy J, Twelves C, Aliman D, et al. Effect of food on the pharmacokinetics of capecitabine and its metabolites following oral administration in cancer patients. Clin Cancer Res. 1998;4(4):941-8.
42. Valle M, Di Salle E, Jannuzzo MG, Poggesi I, Rocchetti M, Spinelli R. A predictive model for exemestane pharmacokinetics/pharmacodynamics incorporating the effect of food and formulation. Br J Clin Pharmacol. 2005 ;59(3):355-64.
43. Sioufi A, Sandrenan N, Godbillon J, Trunet P, Czendlik C, Howald H, et al. Comparative bioavailability of letrozole under fed and fasting conditions in 12 healthy subjects alter a 2.5 mg single oral administration. Biophann Drug Dispos. 1997;18(6):489-97.
44. Deschamps B, Musaji N, Gillespie JA. Food effect on the bioavailability of two distinct formulations of megestrol acetate oral suspension. Int J Nanomedicine. 2009;4:185-92.
45. Cockshott ID. Bicalutamide: clinical pharmacokinetics and metabolism. Clin Pharmacokinet. 2004;43(13):855-78.
46. Gunnarsson PO, Davidsson T, Andersson SB, Backman C, Johansson SA. Impairment of estramustine phosphate absorption by concurrent intake of milk and food. Eur J Clin Pharmacol. 1990;38(2):189-93.
47. Herbes VMM, Rosing H, ten Bokke Huinink WW, van Zomeren DM, Batchelor D, Doyle E. Oral topotecan: bioavailability and effect of food co-administration. Br J Cancer. 1999;80(9):1380-6.
48. Bugat R, Variol P, Roché H, Fumoleau P, Robinet G, Senac I. The effects of food on the pharmacokinetic profile of oral vinorelbine. Cancer Chemother Pharmacol. 2002;50(4):285-90.
49. Laitinen K, Patronen A, Hou P, Lôyttynienii E, Pylkkânen L, Kleimola T, et al. Timing of food intake has a marked effect on the bioavailability of clodronate. Bone. 2000;27(2):293-6.
50. Teoa SK, Schefflera MR, Kookb KA, Tracewellc WG, Colbumc WA, Stirlinga DI, et al. Effect of a high-fat meal on thalidomide pharmacokinetics and the relative bioavailability of oral formulations in healthy men and women. Biopharm Drug Dispos. 2000;21(1):33-40.
51. Rubis EH, Agrawal NGB, Friedman EJ, Scott P, Mazina KE, Sun L, et al. A study to determine the effects of food and multiple dosing on the pharmacokinetics of vorinostat given orally to patients with advanced cancer. Clin Cancer Res. 2006;12(23):7039-45.