|
Abstract: |
Keywords: Fludarabine, stability
Fludarabine is an important drug in therapy of chronic lymphocytic leukaemia [1]. The administration and the knowledge of suitable storage conditions improve safety and can reduce costs. However, knowledge about the stability with up-to-date analytical methods is also important. A detailed collection of data about stability and compatibility of parenterally applicable cytostatic drugs is published on the Stabilis database [2]. The aim of this study was to determine the shelf life of fludarabine phosphate as a concentrated solution 25 mg/mL after opening of the vial, as well as that of the drug compounded in infusion bags. For this, HPLC assays and pH measurements were done at the beginning and end of the stability study.
Materials
To investigate the stability, original vials with 25 mg/mL fludarabine phosphate (Teva, Batch Nr. 07C08NA and 07E29KA) were opened and infusion bags with average concentrations of 0.04 mg/mL, 0.2 mg/mL and 1 mg/mL were prepared. The preparation of the infusion bags was performed using aseptic technique under laminar airflow in a Class II safety cabinet. For the purpose of this experiment these concentrations were assumed as standard concentrations. The infusion bags contained NaCl 0.9% as diluent. The samples were then stored between +2°C and +8°C or +25°C with or without light protection. On days 0, 1, 2, 7, 14 and 21, samples were taken and frozen at -26°C. The samples were measured twice. For checking degradation products a gradient method described in the qualitative validation was used, whereas for the assay the method of the European Pharmacopoeia 7.0 (Ph. Eur. 7.0) [3] was used. PH measurements were performed at d0 and d21 with a laboratory pH meter with integrated printer (inoLab pH Level 2P by WTW). All vials and infusion bags were inspected visually at each sampling.
The polyolefin infusion bags (Macoflex N 100 mL) containing 0.9% sodium chloride were purchased from MacoPharma (Langen, Germany). Methanol was HPLC grade and all other chemicals were at least of analytical grade.
HPLC method
The HPLC system consisted of a Knauer HPLC pump K-1001, autosampler SIL-10ADvp (Shimadzu, Duisburg, Germany), column oven CTO-10ASvp (Shimadzu, Duisburg, Germany), diode array detector Wellchrom DAD K-2700 (Knauer GmbH, Berlin, Germany) and a PC with Software EuroChrom 2000 Version 2.05 (Knauer GmbH, Berlin, Germany). For the stationary phase a reversed-phase column Nucleodur RP18e 5 µm 150 × 4.6 mm (Macherey-Nagel, Düren, Germany) with a precolumn: CC8/4 Nucleosil 100-5 C18 (Macherey-Nagel, Düren, Germany) was used. The temperature was set at 40°C. For the quantitative analysis the isocratic method of the Ph. Eur. 7.0 with a mobile phase 200 volume units methanol and 800 volume units of a solution of potassium dihydrogen phosphate (1.36 g/L) was used. A gradient method with a solution of potassium dihydrogen phosphate (1.36 g/L) and methanol was used to check all related substances (or impurities) mentioned in the Ph. Eur. 7.0. The gradient started at 0 minutes with 6% methanol, rising to 20% at 10 minutes. The flow rate was 1 mL/min. Detection wavelength was 260 nm.
Qualitative method validation
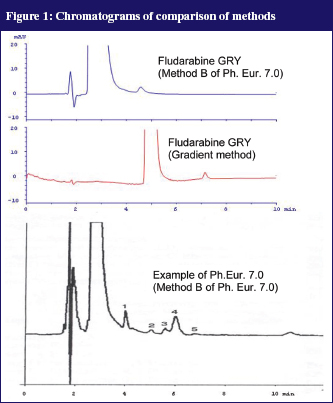
In the Ph. Eur. 7.0 two isocratic HPLC methods for determination of the purity of fludarabine phosphate were described. Method A captures the early eluting (more hydrophilic) compounds, Method B captures only the more lipophilic compounds. As no reference compounds of the impurities could be used, the methods from the Ph. Eur. 7.0 were copied as close as possible. It appeared that the impurities that were shown in the example chromatograms in the Ph. Eur. 7.0 were not found in our own chromatograms. Our ‘in-house’ developed gradient method, see Figure 1, produced a considerably sharper peak but was also not able to make the impurities visible. Thus, it is likely that the impurities were not present in the initial product.
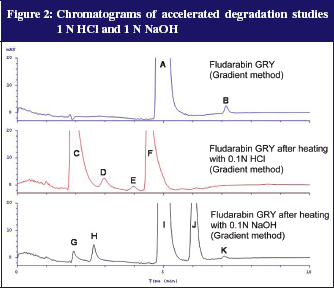
Additionally, degradation tests with 0.1N NaOH and 0.1N HCl were performed. For this, 100 µl fludarabine GRY 25 mg/mL was added to either 900 µl 0.1N NaOH or 0.1N HCl and heated to 80°C for 15 minutes. The samples were subsequently diluted with distilled water to a dilution of 100 µg/mL. The acidic variant corresponds with the preparation of reference solution B of the fludarabine phosphate monograph (Ph. Eur. 7.0) except for the final dilution (100 µl instead of 200 µl). Figure 2 shows the results.
After both accelerated conditions it was possible to detect well separated degradation products. The UV spectra of the peaks give additional information: peaks C, G and H each have a stronger signal at 292 nm compared to the extinction maximum of fludarabine and the measuring wavelength of 260 nm. This fits the description of impurities A and B in the monograph (Ph. Eur. 7.0.) From that, signals C, G and H possibly relate to impurities A and B, described in the Ph. Eur. 7.0. This was a further indication that the gradient method separates all impurities mentioned in the Ph. Eur. 7.0 and that most impurities were not present in fludarabine GRY.
Quantitative method validation
Fludarabine phosphate was calibrated at 260 nm showing linearity in a concentration range from 10 µg/mL to 100 µg/mL (n = 6, all RSD < 2.0%, r = 0.9999). Control Standards were measured after each of the six measurements. The inter-day coefficient of variation over the whole period of measurements was smaller than 3.0%.
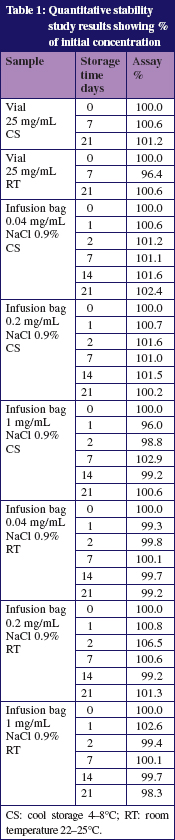
Table 1 shows the results of the assays of the vials. None of the samples showed a change in colour or a precipitation. Degradation products were not detected. The pH which varied between pH 4.8 and pH 6.9 in all infusion bags and vials did not change significantly during the storage.
The described method separates the degradation products under stress with acid and alkaline conditions, furthermore it separates all impurities mentioned in the Ph. Eur. 7.0. In all vials and infusion bags no degradation products were found during the storage time of 21 days.
The stability of fludarabine in standard therapeutically used concentrations was shown for at least 21 days when stored at 8°C or at 25°C if stability is defined as content > 95%, no degradation products, no significant pH change and no visible change.
This research was kindly supported by a grant from GRY Pharma GmbH, Mörfelden, Germany.
Dr rer nat Rainer Trittler, PhD
Department of Pharmacy
University Hospital Freiburg
55 Hugstetterstr
DE-79106 Freiburg, Germany
1. Rai KR, Peterson BL, Appelbaum FR, Kolitz J, Elias L, Sheperd L, et al. Fludarabine compared with chlorambucil as primary therapy for chronic lymphocytic leukemia. N Engl J Med. 2000;343(24):1750-7.
2. Vigneron J. Stabilis. [cited 2012 Feb 16]. Available from: www.stabilis.org
3. Fludarabine Phosphate. European Pharmacopoeia. 7th ed. 2011.
Source URL: https://ppme.i2ct.eu/ejop_article/new-stability-studies-for-fludarabine-according-to-the-european-pharmacopoeia-7-0
Copyright ©2024 PPME unless otherwise noted.